library(readr)
library(ggplot2)
library(dplyr)
library(tidyr)
## theme for ggplot
theme_set(theme_classic())
theme_update(text = element_text(size = 14))Solution for exercise on browsing damage on tree saplings
1 Preparations
- Loading required packages and set ggplot theme.
2 Read the data
dat_browse <- read_csv("data/09_dataset2_tree_browsing.csv")
dat_browse# A tibble: 179 × 4
canopy_closure species N_browsed N_total
<dbl> <chr> <dbl> <dbl>
1 78 Acer 10 23
2 78 Fagus 12 55
3 78 Picea 1 15
4 61 Acer 20 31
5 61 Fagus 6 13
6 61 Picea 3 40
7 69 Acer 19 55
8 69 Fagus 17 41
9 69 Picea 1 46
10 73 Acer 25 50
# ℹ 169 more rows3 Graphical data exploration
We are interested in the proportion of browsed sapling, so we calculate this first:
dat_browse <- dat_browse %>%
mutate(prop_browsed = N_browsed/N_total)Now, we can plot the relationship of tthe two explanatory variables with the proportion of browsed saplings:
ggplot(dat_browse, aes(canopy_closure, prop_browsed, color = species)) +
geom_point() 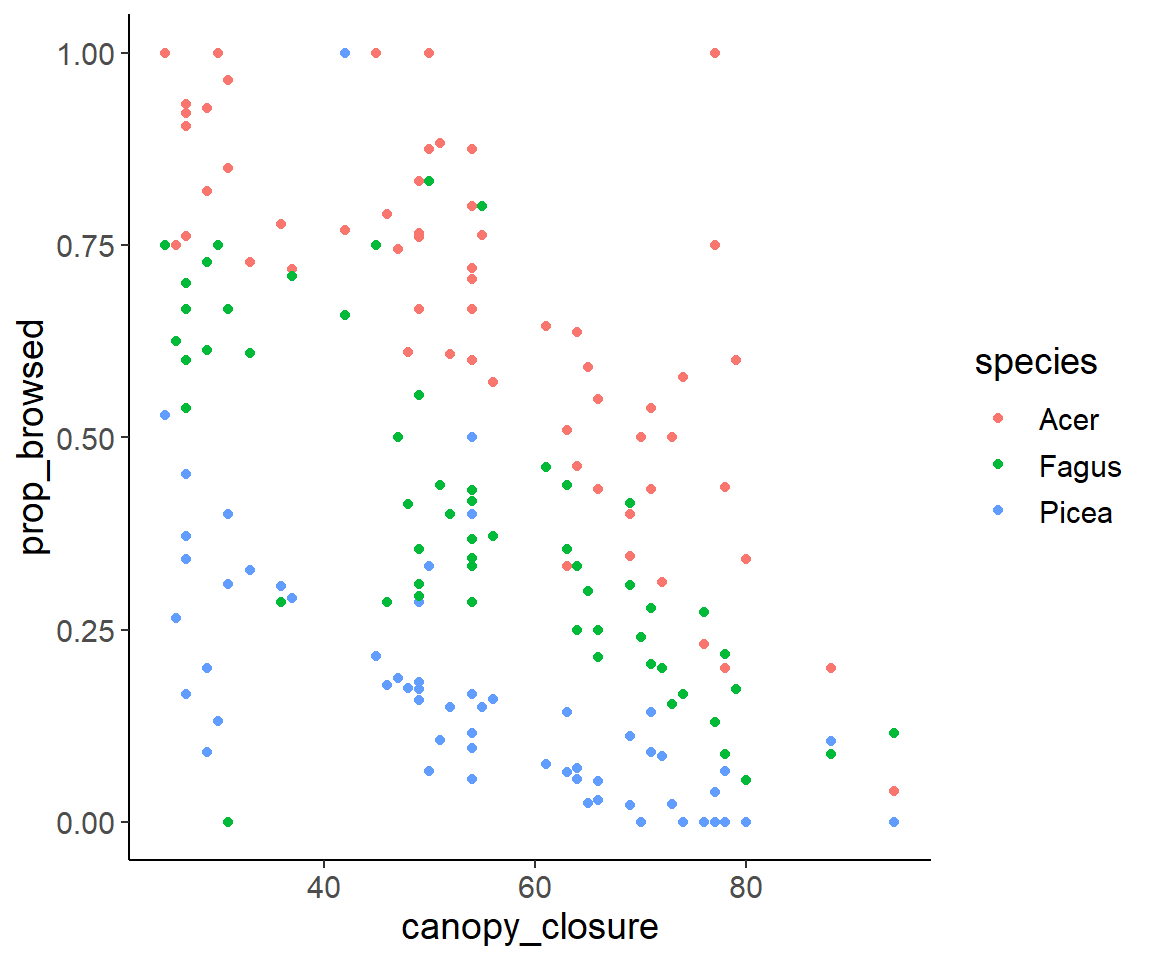
We can “guesstimate” that rowdeer like Acer best and Picea least. Furthermore, browsing might decrease with canopy closure, but of course these guesses should be tested with statistics.
4 Statistical modelling
4.1 Fitting the first model
Since we have proportion data here, we chose a GLM with binomial distribution and logit link function as model. Note the response variable with two columns, which are the no. of browsed sapling and the no. of unbrowsed saplings.
glm1 <- glm(cbind(N_browsed, N_total - N_browsed) ~ canopy_closure*species,
data = dat_browse, family = "binomial")
summary(glm1)
Call:
glm(formula = cbind(N_browsed, N_total - N_browsed) ~ canopy_closure *
species, family = "binomial", data = dat_browse)
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) 3.665267 0.242568 15.110 < 2e-16 ***
canopy_closure -0.054936 0.003972 -13.832 < 2e-16 ***
speciesFagus -1.587928 0.309384 -5.133 2.86e-07 ***
speciesPicea -2.891099 0.327896 -8.817 < 2e-16 ***
canopy_closure:speciesFagus 0.008344 0.005224 1.597 0.110
canopy_closure:speciesPicea 0.003982 0.006236 0.639 0.523
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 1491.08 on 178 degrees of freedom
Residual deviance: 192.27 on 173 degrees of freedom
AIC: 719.83
Number of Fisher Scoring iterations: 4There is no indication of overdispersion (192.27/173 = 1.11) so we do not need to account for it and can test the interaction
test1 <- drop1(glm1, test = "Chi")
test1Single term deletions
Model:
cbind(N_browsed, N_total - N_browsed) ~ canopy_closure * species
Df Deviance AIC LRT Pr(>Chi)
<none> 192.27 719.83
canopy_closure:species 2 194.85 718.41 2.5852 0.2746The interaction is not supported so we also fit a model with the both main effects only.
glm2 <- glm(cbind(N_browsed, N_total - N_browsed) ~ canopy_closure + species,
data = dat_browse, family = "binomial")
test2 <- drop1(glm2, test = "Chi")
test2Single term deletions
Model:
cbind(N_browsed, N_total - N_browsed) ~ canopy_closure + species
Df Deviance AIC LRT Pr(>Chi)
<none> 194.85 718.41
canopy_closure 1 781.51 1303.07 586.66 < 2.2e-16 ***
species 2 1101.65 1621.21 906.80 < 2.2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1The main effects have very strong statistical support. Clearly, this finding will not be changed by p-value adjustment because of multiple testing, but for completeness I show it here again:
4.2 P-value adjustment because of multiple testing
p_vals <- c(test1$`Pr(>Chi)`[2], test2$`Pr(>Chi)`[2:3])
p_vals[1] 2.745551e-01 1.333463e-129 1.230754e-197As expected the two p-values for the main effects are essentially zero. So we have strong evidence that canopy closure has an effect on browsing and species differ in their browsing.
5 Create and plot predictions
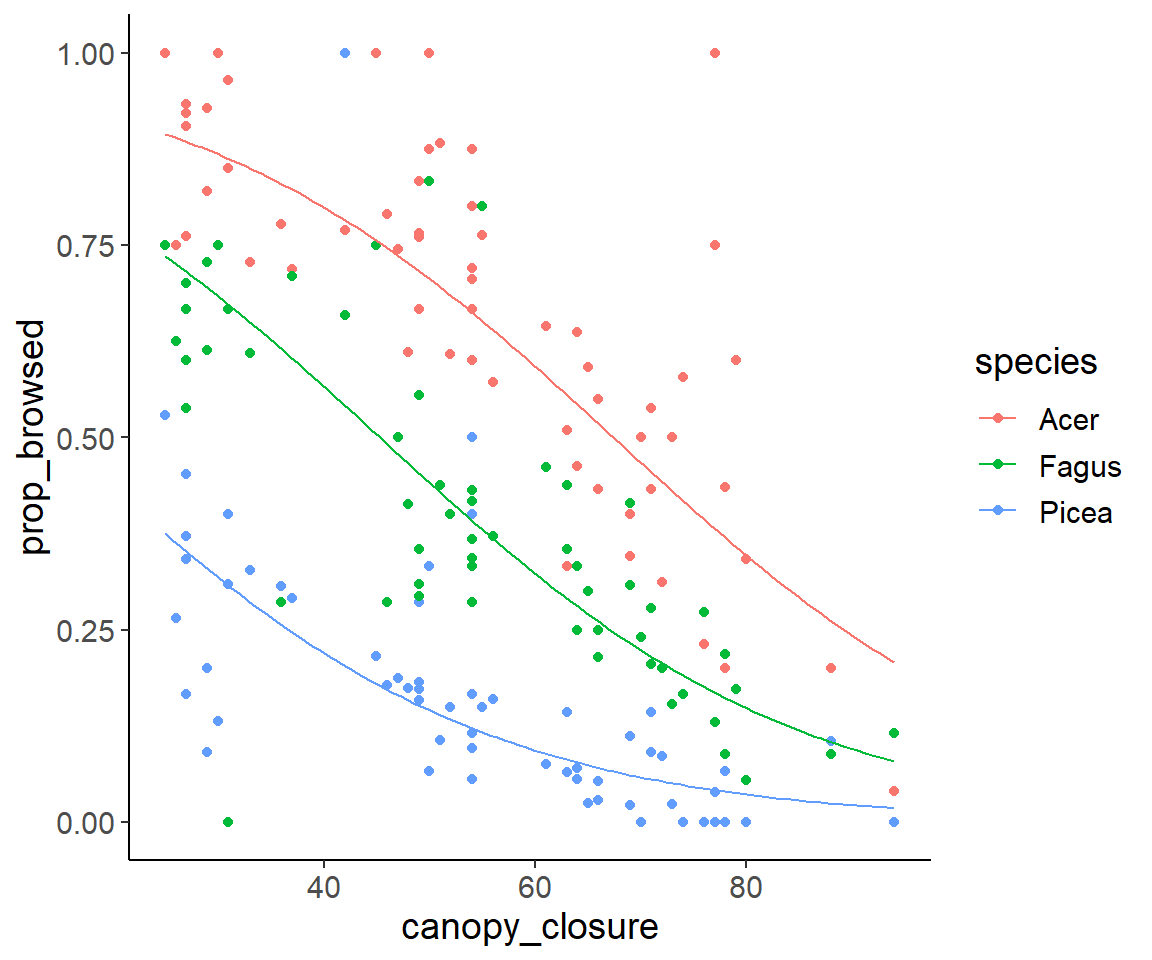
Finally, we plot the data together with the predictions of the model with the two main effects.
As usual, we first generate new data for predictions:
pred_dat <- expand_grid(canopy_closure = seq(min(dat_browse$canopy_closure),
max(dat_browse$canopy_closure),
length = 100),
species = unique(dat_browse$species))
pred_dat$pred_pi <- predict(glm2, pred_dat, type = "response")And then we can plot everything:
ggplot(dat_browse, aes(canopy_closure, prop_browsed, color = species)) +
geom_point() +
geom_line(aes(y = pred_pi), data = pred_dat)